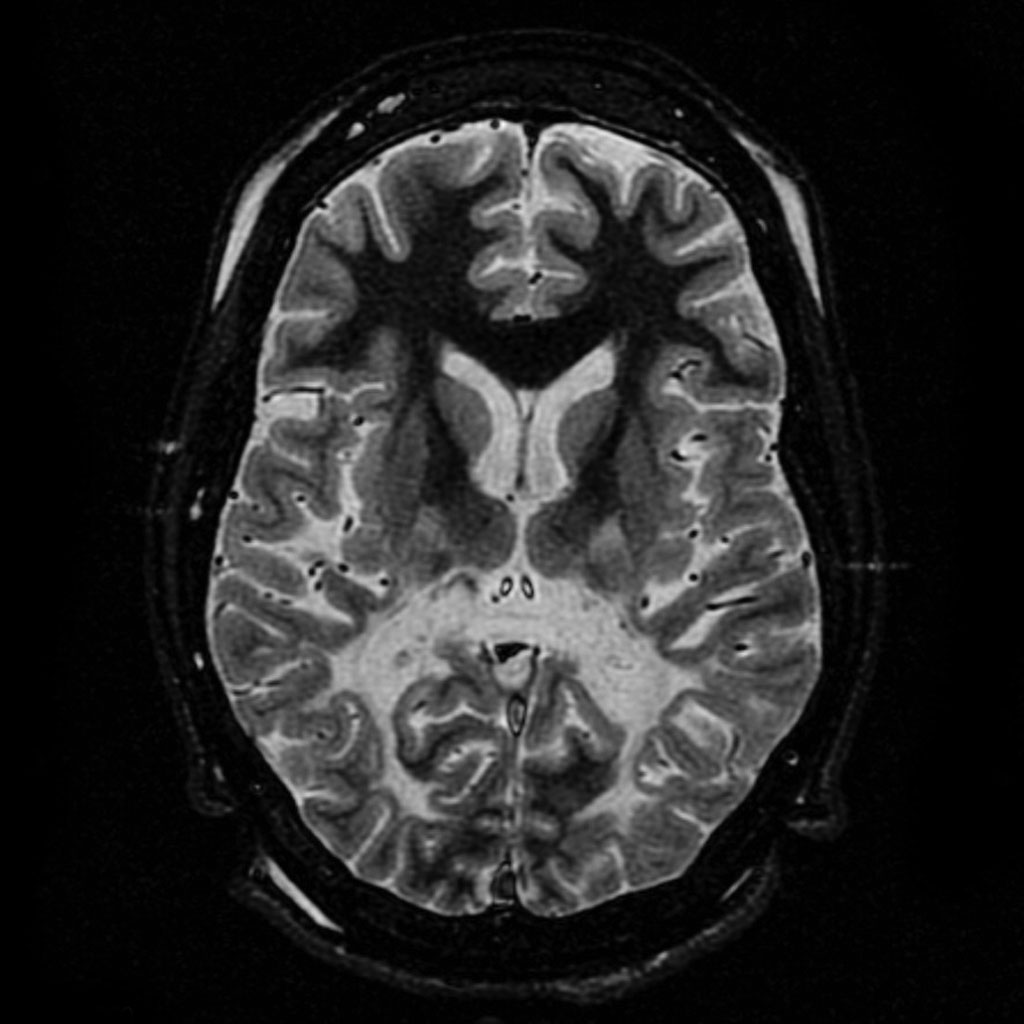
Pengenalan: Misteri Saraf yang Dimakan Lemak
Adrenoleukodystrophy (ALD) mungkin kedengaran seperti nama makhluk asing dari filem fiksyen sains, tetapi ia adalah realiti pahit bagi ribuan keluarga di seluruh dunia. Penyakit ini, yang disebarkan melalui kromosom X, adalah contoh sempurna bagaimana satu kesilapan kecil dalam metabolisme boleh mencetuskan malapetaka neurologi. Bayangkan kanak-kanak yang normal dan ceria tiba-tiba kehilangan keupayaan untuk berjalan, bercakap, dan akhirnya, hidup—semuanya kerana asid lemak rantai panjang (VLCFA) yang tidak dapat diproses. Sebagai penulis sains, saya tidak boleh tidak kagum dengan kerumitan dan kebrutalan penyakit ini.
Biologi Molekul: Kegagalan Peroksisom dan Banjir Asid Lemak
Pada tahap selular, ALD disebabkan oleh kegagalan peroksisom—organel kecil yang bertanggungjawab untuk mengoksidakan asid lemak. Dalam pesakit ALD, terdapat mutasi pada gen ABCD1 yang mengodkan protein pengangkut ke peroksisom. Akibatnya, asid lemak rantai panjang (terutamanya C24:0 dan C26:0) tidak dapat dipecahkan dan terkumpul dalam tisu seluruh badan. Ini adalah fenomena yang menakjubkan dari segi biokimia: satu kecacatan enzimatik boleh menyebabkan kaskade kerosakan yang meluas. VLCFA ini sangat toksik kepada sel-sel saraf, terutama oligodendrosit yang menghasilkan myelin—lapisan pelindung yang membolehkan isyarat elektrik bergerak pantas di sepanjang neuron. Apabila myelin rosak, komunikasi antara otak dan badan menjadi huru-hara.
Patologi: Kerosakan Sistem Saraf Pusat dan Kelenjar Adrenal
Kesan utama ALD adalah pada tiga sistem utama: sistem saraf pusat, korteks adrenal, dan sel Leydig dalam testis. Dalam otak, pengumpulan VLCFA menyebabkan demielinasi progresif—myelin hancur seperti kabel elektrik yang terdedah. Ini menyebabkan sawan, hiperaktif, masalah pertuturan, dan gangguan kognitif. Paling mengejutkan, penyakit ini boleh muncul dalam pelbagai bentuk: kanak-kanak lelaki boleh mengalami bentuk serebral kanak-kanak yang agresif, manakala dewasa mungkin hanya menunjukkan gejala ringan seperti keletihan atau disfungsi adrenal. Kelenjar adrenal pula rosak oleh deposit lemak, menyebabkan kekurangan kortisol dan aldosteron—situasi yang boleh membawa maut jika tidak dirawat. Pada lelaki, testis juga terjejas, menyebabkan masalah kesuburan.
Varian Klinikal: Kepelbagaian Fenotip yang Membingungkan
Salah satu aspek paling menarik ALD adalah heterogeniti klinikalnya. Walaupun gen yang sama bermutasi, gejala boleh berbeza drastik antara individu. Kira-kira satu pertiga pesakit lelaki akan mengalami bentuk serebral kanak-kanak yang paling teruk, dengan penurunan pesat dalam fungsi neurologi dalam tempoh 2-5 tahun selepas diagnosis. Selebihnya mungkin mengalami adrenomieloneuropati (AMN) yang lebih ringan, hanya melibatkan saraf tunjang dan saraf periferi. Lebih menarik, 80% wanita heterozigot—pembawa gen—juga boleh menunjukkan gejala ringan seperti kelemahan kaki atau masalah kencing, walaupun pada usia lanjut. Ini menunjukkan bahawa ALD bukan sekadar penyakit lelaki, tetapi juga ancaman tersembunyi untuk wanita.
Diagnostik dan Rawatan: Cabaran dan Harapan
Diagnosis ALD kini boleh dilakukan melalui ujian darah untuk mengukur tahap VLCFA, dan disahkan dengan ujian genetik. Namun, rawatan masih terhad. Terapi utama adalah pemindahan sumsum tulang atau terapi gen, yang boleh menghentikan perkembangan penyakit jika dilakukan awal. Minyak Lorenzo—campuran asid oleik dan erusik—pernah dianggap sebagai penyelamat, tetapi kini terbukti hanya berkesan dalam mengurangkan gejala ringan. Satu penemuan menakjubkan adalah bahawa terapi gen menggunakan virus lentiviral untuk memperkenalkan gen ABCD1 yang sihat telah menunjukkan kejayaan dalam ujian klinikal, memberikan harapan baru. Selain itu, pemantauan rapi fungsi adrenal dan terapi penggantian hormon boleh menyelamatkan nyawa pesakit dengan kekurangan adrenal.
Kesimpulan: Perjuangan Melawan Masa dan Lemak
Adrenoleukodystrophy adalah peringatan keras tentang betapa rapuhnya tubuh manusia. Walaupun ia adalah penyakit jarang, impaknya terhadap keluarga dan pesakit adalah dahsyat. Namun, di sebalik tragedi ini, sains terus maju. Dari pemahaman asas tentang peroksisom hingga ke terapi gen yang revolusioner, kita menyaksikan bagaimana penyelidikan boleh mengubah nasib. Sebagai penulis sains, saya kagum dengan ketabahan pesakit dan dedikasi saintis yang berjuang menentang masa. ALD mungkin memakan otak, tetapi ia tidak dapat memakan semangat manusia untuk mencari penawar.
---
Rujukan: Adrenoleukodystrophy — Wikipedia
Adrenoleukodystrophy: Saat Lemak Memakan Otak Manusia Sendiri. Bayangkan tubuh anda mula membina asid lemak rantai panjang secara berlebihan, merosakkan saraf dan otak—itulah realiti pesakit Adrenoleukodystrophy. Penyakit genetik ini, yang terpaut pada kromosom X, boleh membawa maut dalam masa beberapa tahun selepas simptom muncul. Temui bagaimana mekanisme misteri ini berfungsi dan apa yang saintis lakukan untuk melawannya.. Pengenalan: Misteri Saraf yang Dimakan Lemak
Adrenoleukodystrophy ALD mungkin kedengaran seperti nama makhluk asing dari filem fiksyen sains, tetapi ia adalah realiti pahit bagi ribuan keluarga di seluruh dunia. Penyakit ini, yang disebarkan melalui kromosom X, adalah contoh sempurna bagaimana satu kesilapan kecil dalam metabolisme boleh mencetuskan malapetaka neurologi. Bayangkan kanak-kanak yang normal dan ceria tiba-tiba kehilangan keupayaan untuk berjalan, bercakap, dan akhirnya, hidup—semuanya kerana asid lemak rantai panjang VLCFA yang tidak dapat diproses. Sebagai penulis sains, saya tidak boleh tidak kagum dengan kerumitan dan kebrutalan penyakit ini.
Biologi Molekul: Kegagalan Peroksisom dan Banjir Asid Lemak
Pada tahap selular, ALD disebabkan oleh kegagalan peroksisom—organel kecil yang bertanggungjawab untuk mengoksidakan asid lemak. Dalam pesakit ALD, terdapat mutasi pada gen ABCD1 yang mengodkan protein pengangkut ke peroksisom. Akibatnya, asid lemak rantai panjang terutamanya C24:0 dan C26:0 tidak dapat dipecahkan dan terkumpul dalam tisu seluruh badan. Ini adalah fenomena yang menakjubkan dari segi biokimia: satu kecacatan enzimatik boleh menyebabkan kaskade kerosakan yang meluas. VLCFA ini sangat toksik kepada sel-sel saraf, terutama oligodendrosit yang menghasilkan myelin—lapisan pelindung yang membolehkan isyarat elektrik bergerak pantas di sepanjang neuron. Apabila myelin rosak, komunikasi antara otak dan badan menjadi huru-hara.
Patologi: Kerosakan Sistem Saraf Pusat dan Kelenjar Adrenal
Kesan utama ALD adalah pada tiga sistem utama: sistem saraf pusat, korteks adrenal, dan sel Leydig dalam testis. Dalam otak, pengumpulan VLCFA menyebabkan demielinasi progresif—myelin hancur seperti kabel elektrik yang terdedah. Ini menyebabkan sawan, hiperaktif, masalah pertuturan, dan gangguan kognitif. Paling mengejutkan, penyakit ini boleh muncul dalam pelbagai bentuk: kanak-kanak lelaki boleh mengalami bentuk serebral kanak-kanak yang agresif, manakala dewasa mungkin hanya menunjukkan gejala ringan seperti keletihan atau disfungsi adrenal. Kelenjar adrenal pula rosak oleh deposit lemak, menyebabkan kekurangan kortisol dan aldosteron—situasi yang boleh membawa maut jika tidak dirawat. Pada lelaki, testis juga terjejas, menyebabkan masalah kesuburan.
Varian Klinikal: Kepelbagaian Fenotip yang Membingungkan
Salah satu aspek paling menarik ALD adalah heterogeniti klinikalnya. Walaupun gen yang sama bermutasi, gejala boleh berbeza drastik antara individu. Kira-kira satu pertiga pesakit lelaki akan mengalami bentuk serebral kanak-kanak yang paling teruk, dengan penurunan pesat dalam fungsi neurologi dalam tempoh 2-5 tahun selepas diagnosis. Selebihnya mungkin mengalami adrenomieloneuropati AMN yang lebih ringan, hanya melibatkan saraf tunjang dan saraf periferi. Lebih menarik, 80% wanita heterozigot—pembawa gen—juga boleh menunjukkan gejala ringan seperti kelemahan kaki atau masalah kencing, walaupun pada usia lanjut. Ini menunjukkan bahawa ALD bukan sekadar penyakit lelaki, tetapi juga ancaman tersembunyi untuk wanita.
Diagnostik dan Rawatan: Cabaran dan Harapan
Diagnosis ALD kini boleh dilakukan melalui ujian darah untuk mengukur tahap VLCFA, dan disahkan dengan ujian genetik. Namun, rawatan masih terhad. Terapi utama adalah pemindahan sumsum tulang atau terapi gen, yang boleh menghentikan perkembangan penyakit jika dilakukan awal. Minyak Lorenzo—campuran asid oleik dan erusik—pernah dianggap sebagai penyelamat, tetapi kini terbukti hanya berkesan dalam mengurangkan gejala ringan. Satu penemuan menakjubkan adalah bahawa terapi gen menggunakan virus lentiviral untuk memperkenalkan gen ABCD1 yang sihat telah menunjukkan kejayaan dalam ujian klinikal, memberikan harapan baru. Selain itu, pemantauan rapi fungsi adrenal dan terapi penggantian hormon boleh menyelamatkan nyawa pesakit dengan kekurangan adrenal.
Kesimpulan: Perjuangan Melawan Masa dan Lemak
Adrenoleukodystrophy adalah peringatan keras tentang betapa rapuhnya tubuh manusia. Walaupun ia adalah penyakit jarang, impaknya terhadap keluarga dan pesakit adalah dahsyat. Namun, di sebalik tragedi ini, sains terus maju. Dari pemahaman asas tentang peroksisom hingga ke terapi gen yang revolusioner, kita menyaksikan bagaimana penyelidikan boleh mengubah nasib. Sebagai penulis sains, saya kagum dengan ketabahan pesakit dan dedikasi saintis yang berjuang menentang masa. ALD mungkin memakan otak, tetapi ia tidak dapat memakan semangat manusia untuk mencari penawar.
---
Rujukan: Adrenoleukodystrophy — Wikipedia https://en.wikipedia.org/wiki/Adrenoleukodystrophy