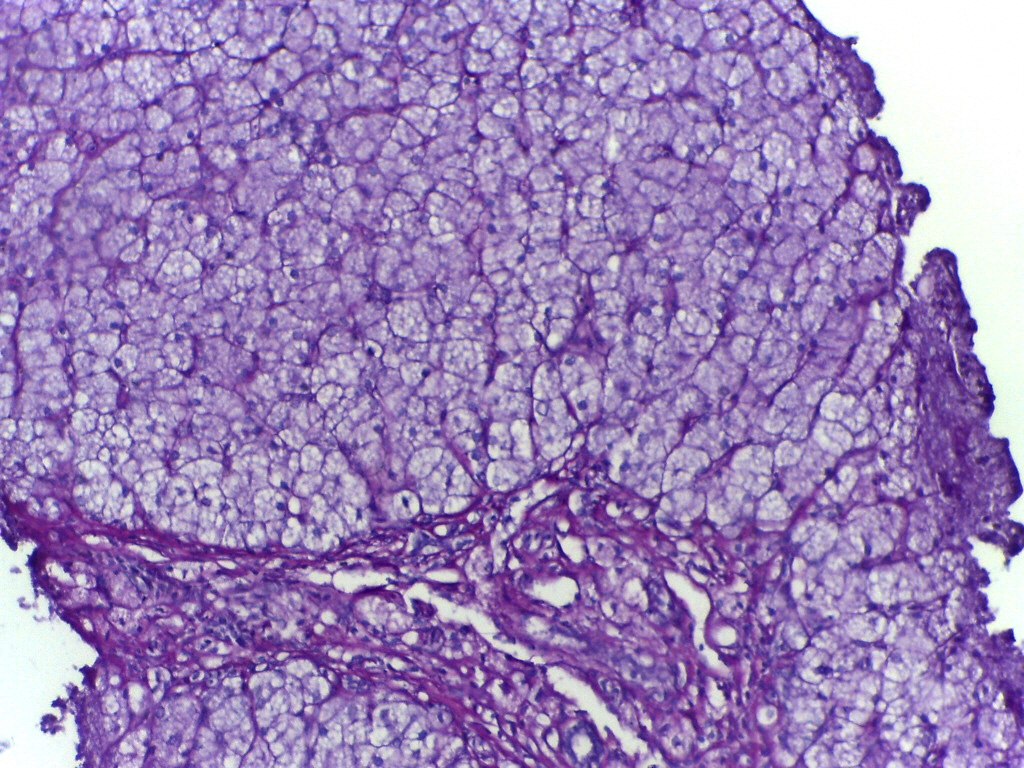
¿Qué es realmente la 'enfermedad de almacenamiento de glucógeno' y por qué no es simplemente un 'problema de azúcar'?
Imagina que el glucógeno es una batería de respaldo en el cuerpo: se almacena principalmente en el hígado y los músculos, y se libera como glucosa cuando se necesita energía — por ejemplo, durante la noche, ayuno breve o actividad física. En pacientes con Enfermedad de Almacenamiento de Glucógeno (GSD), este sistema
está dañado a nivel molecular. No es porque coman demasiado azúcar, ni por estilo de vida — sino porque un enzima o proteína transportadora específica
no existe en absoluto, o funciona parcialmente. Como resultado: el glucógeno se acumula en exceso en el hígado/músculo como 'residuos químicos', o no se puede construir directamente. No es diabetes. No es hipoglucemia común. Es un fallo en la máquina bioquímica hereditaria — y hay más de 16 tipos de GSD reconocidos científicamente, cada uno causado por mutaciones genéticas específicas.
¿Por qué los bebés parecen sanos en la primera semana y luego colapsan después de 4 horas de ayuno?
La mayoría de las GSD (especialmente los tipos I, III, VI, IX) muestran los primeros síntomas entre el día 3 y el mes 2 de vida —
cuando los depósitos de glucosa prenatal se agotan. Los recién nacidos aún pueden depender de la glucosa del cordón umbilical; pero una vez nacidos, dependen del glucógeno hepático. En el tipo I de GSD (von Gierke), el enzima glucosa-6-fosfatasa no está presente. Por lo tanto, aunque el hígado esté lleno de glucógeno,
no puede liberar glucosa a la sangre. Como resultado: hipoglucemia grave en 3–4 horas sin leche. Síntomas: debilidad, palidez, convulsiones — y si no se trata inmediatamente, coma metabólico en menos de 24 horas. Un estudio en el Hospital Kuala Lumpur (2022) registró 7 casos de GSD-I no diagnosticados hasta los 18 meses — todos ingresaron en la UCI debido a hipoglucemia de causa desconocida.
¿Por qué las pruebas de sangre normales a menudo fallan en detectar GSD y qué pruebas deben realizarse realmente?
Muchos médicos sospechan infección o epilepsia cuando un bebé tiene convulsiones sin fiebre. Las pruebas básicas de sangre (glucosa, electrolitos, función hepática) muestran efectivamente hipoglucemia + aumento de lactato + triglicéridos altos — pero esto también ocurre en sepsis o fallo hepático agudo. Señal única de GSD:
hipoglucemia con lactato alto Y acidosis metabólica QUE NO MEJORA tras la administración de glucosa intravenosa. Si la glucosa IV mejora rápidamente la hipoglucemia, probablemente no sea GSD. Si no, es una fuerte señal de fallo en la vía de gluconeogénesis. Prueba definitiva: análisis de enzimas en biopsia hepática (estándar de oro), o secuenciación genética
G6PC (para GSD-Ia),
AGL (para GSD-III) o
PHKA2 (para GSD-IX). Ahora, las pruebas genéticas de panel GSD se pueden realizar en 10 días — sin biopsia — en el Centro Nacional de Genética UKM.
¿Puede curarse la GSD? ¿Y por qué la dieta alta en maíz no es un mito, sino un protocolo diario obligatorio?
No hay tratamiento genético ni medicamento mágico — pero la GSD
se puede controlar completamente con manejo nutricional preciso. Para el tipo I de GSD, la estrategia principal es
prevenir la hipoglucemia manteniendo un flujo constante de glucosa. Cómo: comer cada 2–3 horas durante el día y la noche — usando
almidón crudo de maíz (UCCS). Este almidón se digiere lentamente, proporcionando glucosa estable durante 6–8 horas. Un estudio longitudinal de 20 años en Singapur mostró que el 94% de los pacientes con GSD-I que siguieron el UCCS desde bebés nunca sufrieron coma metabólico — y alcanzaron crecimiento y logros académicos normales. Importante: el UCCS
no es un suplemento, sino un medicamento. La dosis debe calcularse según el peso y la edad — y
debe iniciarse bajo la supervisión de un especialista en metabolismo, no por recomendación de una tía en WhatsApp.
¿Por qué la GSD no es solo una 'enfermedad infantil' y cuáles son los riesgos a largo plazo que a menudo se ignoran?
Mucha gente piensa que la GSD 'desaparece' cuando el niño crece. Gran error. Sin manejo de por vida, complicaciones surgen: hígado graso progresivo → cirrosis → cáncer hepático (riesgo 10 veces mayor a los 30 años); aumento de ácido úrico → gota y cálculos renales; hiperlipidemia → aterosclerosis temprana. En el tipo II de GSD (enfermedad de Pompe), la debilidad muscular progresiva puede llevar a insuficiencia respiratoria — y ahora hay enzimas sustitutivas (alglucosidasa alfa) que pueden prolongar la vida 10–15 años si se administran antes de los síntomas clínicos. Lo más crítico:
el screening neonatal para GSD no se incluye en el Programa de Screening Neonatal Nacional de Malasia — aunque se puede detectar mediante espectrometría de masas tandem en la sangre del talón en el día 3. En Alemania y Japón, este screening ha reducido la mortalidad de GSD-I en un 78% en la última década.
¿Cómo pueden actuar las familias y por qué 'esperar a que aparezcan los síntomas' es la decisión más riesgosa?
Si hay antecedentes familiares de GSD, o el bebé muestra: (1) hepatomegalia sin causa clara, (2) hipoglucemia recurrente antes de los 6 meses, (3) retraso en el desarrollo motor acompañado de debilidad muscular —
no esperes. Consulta inmediatamente a un Especialista en Genética o Especialista en Metabolismo Infantil. En Malasia, el Centro Médico de la Universidad de Malaya (PPUM), el Hospital Sultanah Bahiyah Alor Setar y el Instituto de Medicina Genética USM ofrecen consultas y pruebas genéticas certificadas. También hay el Club de Apoyo GSD de Malasia — una comunidad de padres que comparten protocolos alimenticios, consejos sobre UCCS y apoyo emocional 24/7. Recuerda: la GSD no es un castigo. Es un diagnóstico — y un diagnóstico correcto, en el momento adecuado, es el comienzo de una vida plena, activa y larga. Como dijo un paciente con GSD-III de 32 años en Johor: *‘No vivo con la GSD. Vivo
a pesar de la GSD — y ya he corrido dos maratones.’*
---
Réferencia: Enfermedad de almacenamiento de glucógeno — Wikipedia
¿Por qué este niño no puede ayunar más de 4 horas — aunque solo tenga 2 años?. Detrás de su cara dulce y tranquila, su cuerpo alberga una 'bomba de carbohidratos': glucógeno que no se puede descomponer. No hay síntomas claros al nacer, pero el hambre de 4 horas puede provocar convulsiones, coma o muerte súbita. Esto no es un mito. Es una enfermedad rara que afecta a 1 de cada 20.000 bebés — y en 9 de cada 10 casos, no se diagnostica correctamente antes de los 5 años.. ¿Qué es realmente la 'enfermedad de almacenamiento de glucógeno' y por qué no es simplemente un 'problema de azúcar'?
Imagina que el glucógeno es una batería de respaldo en el cuerpo: se almacena principalmente en el hígado y los músculos, y se libera como glucosa cuando se necesita energía — por ejemplo, durante la noche, ayuno breve o actividad física. En pacientes con Enfermedad de Almacenamiento de Glucógeno GSD , este sistema está dañado a nivel molecular . No es porque coman demasiado azúcar, ni por estilo de vida — sino porque un enzima o proteína transportadora específica no existe en absoluto , o funciona parcialmente. Como resultado: el glucógeno se acumula en exceso en el hígado/músculo como 'residuos químicos', o no se puede construir directamente. No es diabetes. No es hipoglucemia común. Es un fallo en la máquina bioquímica hereditaria — y hay más de 16 tipos de GSD reconocidos científicamente, cada uno causado por mutaciones genéticas específicas.
¿Por qué los bebés parecen sanos en la primera semana y luego colapsan después de 4 horas de ayuno?
La mayoría de las GSD especialmente los tipos I, III, VI, IX muestran los primeros síntomas entre el día 3 y el mes 2 de vida — cuando los depósitos de glucosa prenatal se agotan . Los recién nacidos aún pueden depender de la glucosa del cordón umbilical; pero una vez nacidos, dependen del glucógeno hepático. En el tipo I de GSD von Gierke , el enzima glucosa-6-fosfatasa no está presente. Por lo tanto, aunque el hígado esté lleno de glucógeno, no puede liberar glucosa a la sangre . Como resultado: hipoglucemia grave en 3–4 horas sin leche. Síntomas: debilidad, palidez, convulsiones — y si no se trata inmediatamente, coma metabólico en menos de 24 horas. Un estudio en el Hospital Kuala Lumpur 2022 registró 7 casos de GSD-I no diagnosticados hasta los 18 meses — todos ingresaron en la UCI debido a hipoglucemia de causa desconocida.
¿Por qué las pruebas de sangre normales a menudo fallan en detectar GSD y qué pruebas deben realizarse realmente?
Muchos médicos sospechan infección o epilepsia cuando un bebé tiene convulsiones sin fiebre. Las pruebas básicas de sangre glucosa, electrolitos, función hepática muestran efectivamente hipoglucemia + aumento de lactato + triglicéridos altos — pero esto también ocurre en sepsis o fallo hepático agudo. Señal única de GSD: hipoglucemia con lactato alto Y acidosis metabólica QUE NO MEJORA tras la administración de glucosa intravenosa . Si la glucosa IV mejora rápidamente la hipoglucemia, probablemente no sea GSD. Si no, es una fuerte señal de fallo en la vía de gluconeogénesis. Prueba definitiva: análisis de enzimas en biopsia hepática estándar de oro , o secuenciación genética G6PC para GSD-Ia , AGL para GSD-III o PHKA2 para GSD-IX . Ahora, las pruebas genéticas de panel GSD se pueden realizar en 10 días — sin biopsia — en el Centro Nacional de Genética UKM.
¿Puede curarse la GSD? ¿Y por qué la dieta alta en maíz no es un mito, sino un protocolo diario obligatorio?
No hay tratamiento genético ni medicamento mágico — pero la GSD se puede controlar completamente con manejo nutricional preciso. Para el tipo I de GSD, la estrategia principal es prevenir la hipoglucemia manteniendo un flujo constante de glucosa . Cómo: comer cada 2–3 horas durante el día y la noche — usando almidón crudo de maíz UCCS . Este almidón se digiere lentamente, proporcionando glucosa estable durante 6–8 horas. Un estudio longitudinal de 20 años en Singapur mostró que el 94% de los pacientes con GSD-I que siguieron el UCCS desde bebés nunca sufrieron coma metabólico — y alcanzaron crecimiento y logros académicos normales. Importante: el UCCS no es un suplemento , sino un medicamento. La dosis debe calcularse según el peso y la edad — y debe iniciarse bajo la supervisión de un especialista en metabolismo , no por recomendación de una tía en WhatsApp.
¿Por qué la GSD no es solo una 'enfermedad infantil' y cuáles son los riesgos a largo plazo que a menudo se ignoran?
Mucha gente piensa que la GSD 'desaparece' cuando el niño crece. Gran error. Sin manejo de por vida, complicaciones surgen: hígado graso progresivo → cirrosis → cáncer hepático riesgo 10 veces mayor a los 30 años ; aumento de ácido úrico → gota y cálculos renales; hiperlipidemia → aterosclerosis temprana. En el tipo II de GSD enfermedad de Pompe , la debilidad muscular progresiva puede llevar a insuficiencia respiratoria — y ahora hay enzimas sustitutivas alglucosidasa alfa que pueden prolongar la vida 10–15 años si se administran antes de los síntomas clínicos. Lo más crítico: el screening neonatal para GSD no se incluye en el Programa de Screening Neonatal Nacional de Malasia — aunque se puede detectar mediante espectrometría de masas tandem en la sangre del talón en el día 3. En Alemania y Japón, este screening ha reducido la mortalidad de GSD-I en un 78% en la última década.
¿Cómo pueden actuar las familias y por qué 'esperar a que aparezcan los síntomas' es la decisión más riesgosa?
Si hay antecedentes familiares de GSD, o el bebé muestra: 1 hepatomegalia sin causa clara, 2 hipoglucemia recurrente antes de los 6 meses, 3 retraso en el desarrollo motor acompañado de debilidad muscular — no esperes . Consulta inmediatamente a un Especialista en Genética o Especialista en Metabolismo Infantil. En Malasia, el Centro Médico de la Universidad de Malaya PPUM , el Hospital Sultanah Bahiyah Alor Setar y el Instituto de Medicina Genética USM ofrecen consultas y pruebas genéticas certificadas. También hay el Club de Apoyo GSD de Malasia — una comunidad de padres que comparten protocolos alimenticios, consejos sobre UCCS y apoyo emocional 24/7. Recuerda: la GSD no es un castigo. Es un diagnóstico — y un diagnóstico correcto, en el momento adecuado, es el comienzo de una vida plena, activa y larga. Como dijo un paciente con GSD-III de 32 años en Johor: ‘No vivo con la GSD. Vivo a pesar de la GSD — y ya he corrido dos maratones.’
---
Réferencia: Enfermedad de almacenamiento de glucógeno — Wikipedia https://en.wikipedia.org/wiki/Glycogen storage disease