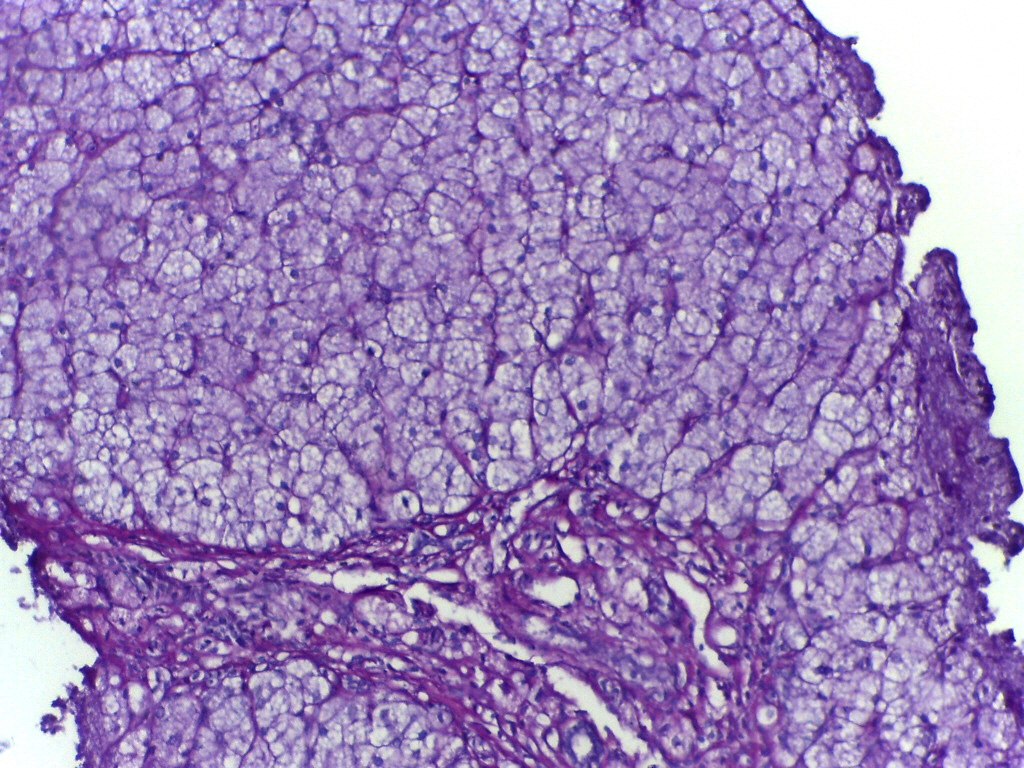
O que é exatamente 'doença de armazenamento de glicogênio' — e por que não é apenas 'problema de açúcar'?
Imagine o glicogênio como uma bateria de reserva no corpo: armazenado principalmente no fígado e nos músculos, e liberado como glicose quando a energia é necessária — por exemplo, durante o sono noturno, jejum breve ou atividade física. Nos pacientes com Doença de Armazenamento de Glicogênio (GSD), o sistema é
dano no nível molecular. Não porque se come muito açúcar, não porque o estilo de vida — mas porque uma enzima ou proteína transportadora específica
não existe ou funciona pela metade. Como resultado: glicogênio acumula-se excessivamente no fígado/músculos como 'lixo químico' — ou, por outro lado, não pode ser construído diretamente. Não é diabetes. Não é hipoglicemia comum. É uma falha de máquina bioquímica congênita — e há mais de 16 tipos de GSD que são cientificamente reconhecidos, cada um causado por uma mutação genética específica.
Por que bebês parecem saudáveis na primeira semana — e então colapsam após 4 horas de jejum?
A maioria dos GSD (especialmente tipos I, III, VI, IX) apresenta os primeiros sintomas entre os dias 3 e 2 meses de vida —
quando a reserva de glicose pré-natal se esgota. Bebês recém-nascidos ainda podem se alimentar de glicose da placenta; mas assim que nascem, eles dependem de glicogênio do fígado. No GSD tipo I (von Gierke), a enzima glukose-6-fosfatoase não existe. Portanto, mesmo que o fígado esteja cheio de glicogênio,
não pode liberar glicose para o fluxo sanguíneo. Como resultado: hipoglicemia grave em 3-4 horas sem leite. Seus sintomas? Lassidão, fraqueza, azulão, convulsões — e se não for tratado imediatamente, coma metabólico em menos de 24 horas. Uma pesquisa no Hospital Kuala Lumpur (2022) registrou 7 casos de GSD-I não diagnosticados até os 18 meses — todos entraram no ICU por hipoglicemia sem causa conhecida.
Por que os testes de sangue comuns frequentemente falham em detectar GSD — e quais testes são reais que devem ser feitos?
Muitos médicos suspeitam de infecção ou epilepsia quando bebês têm convulsões sem febre. Os testes de sangue básicos (glicose, eletrolitos, função do fígado) mostram hipoglicemia + aumento de látex + triglicerídeos altos — mas isso também ocorre em sepsis ou falha hepática aguda. O sinal único do GSD?
Hipoglicemia com látex alto E ASIDOSA METABÓLICA QUE NÃO MELHORA após a administração de glicose intravenosa. Se a glicose IV melhorar a hipoglicemia — é provável que não seja GSD. Se não, é um forte sinal de falha da via de gliconeogênese. O teste conclusivo: análise de enzimas em biópsia de fígado (padrão ouro), ou sequenciamento do gen
G6PC (para GSD-Ia),
AGL (para GSD-III), ou
PHKA2 (para GSD-IX). Agora, o painel de genética do GSD pode ser realizado em 10 dias — sem biópsia — no Centro Genético Nacional UKM.
O GSD pode ser 'curado'? E por que a dieta rica em milho não é mito — mas protocolo diário obrigatório?
Nenhum tratamento genético ou remédio mágico — mas o GSD
pode ser controlado completamente com gerenciamento nutricional preciso. Para o GSD-I, a estratégia principal é
prevenir hipoglicemia mantendo a corrente de glicose contínua. Como? Comer a cada 2-3 horas durante o dia e à noite — usando
amido cru de milho não cozido (UCCS). Esse amido é digerido lentamente, fornecendo glicose estável por 6-8 horas. Uma pesquisa longitudinal de 20 anos na Singapura mostrou que 94% dos pacientes com GSD-I que seguiram UCCS desde bebês nunca tiveram coma metabólico — e alcançaram crescimento e desempenho acadêmico normais. Importante: o UCCS
não é suplemento, mas medicamento. A dose deve ser calculada com base no peso e idade — e
deve ser iniciada sob supervisão de um especialista em metabolismo, não por sugestão de uma tia no WhatsApp.
Por que o GSD não é apenas 'doença de criança' — e quais riscos de longo prazo são frequentemente ignorados?
Muitos pensam que o GSD 'desaparece' quando o filho cresce. Erro grave. Sem gerenciamento a vida inteira, complicações surgem: fígado gorduroso progressivo → sirose → câncer de fígado (risco 10x maior aos 30 anos); aumento de ácido úrico → gota & cálculos renais; hiperlipidemia → aterosclerose precoce. No GSD-II (doença de Pompe), a fraqueza muscular progressiva pode levar ao insuficiência respiratória — e agora há uma enzima substituta (alglucosidase alfa) que pode prolongar a vida em 10-15 anos se for administrada antes dos sintomas clínicos. O mais crítico:
a triagem neonatal para GSD ainda não foi incluída no Programa de Triagem Neonatal Nacional da Malásia — embora possa ser detectada por espectrometria de massa tandem no terceiro dia de sangue de corda. Na Alemanha e no Japão, essa triagem reduziu a mortalidade de GSD-I em 78% nas últimas décadas.
Como as famílias podem agir — e por que 'esperar por sintomas claros' é a decisão mais arriscada?
Se há história familiar de GSD, ou o bebê apresenta: (1) hepatomegalia sem causa clara, (2) hipoglicemia recorrente antes dos 6 meses, (3) atraso do desenvolvimento motor associado à fraqueza muscular —
não espere. Consulte imediatamente um Genetista ou um Metabolista de Crianças. Na Malásia, o Hospital Universitário de Malaca (PPUM), o Hospital Sultanah Bahiyah Alor Setar, e o Instituto de Medicina Genética da USM oferecem consultas e testes genéticos licenciados. Há também o Clube de Apoio ao GSD da Malásia — uma comunidade de pais que compartilha protocolos de alimentação, dicas de UCCS e apoio emocional 24 horas. Lembre-se: o GSD não é uma punição. É um diagnóstico — e um diagnóstico preciso, no momento certo, é o início de uma vida cheia, ativa e longa. Como disse um paciente de 32 anos com GSD-III de Johor: *‘Eu não vivo com GSD. Eu vivo
mesmo com GSD — e eu já corri duas maratonas.’*
---
Rreferência: Doença de armazenamento de glicogênio — Wikipédia
Porquê esse bebê não pode jejar mais de 4 horas — mesmo com apenas 2 anos?. Por trás da aparência alegre e tranquila, o corpo armazena 'bomba de tempo' de carboidratos: glicogênio que não pode ser quebrado. Nenhum sinal claro no nascimento — mas 4 horas de jejum podem desencadear convulsões, coma ou morte súbita. Isso não é mito. Isso é uma doença rara que ataca 1 em cada 20.000 bebês — e 9 de cada 10 casos nunca foram diagnosticados corretamente antes dos 5 anos.. O que é exatamente 'doença de armazenamento de glicogênio' — e por que não é apenas 'problema de açúcar'?
Imagine o glicogênio como uma bateria de reserva no corpo: armazenado principalmente no fígado e nos músculos, e liberado como glicose quando a energia é necessária — por exemplo, durante o sono noturno, jejum breve ou atividade física. Nos pacientes com Doença de Armazenamento de Glicogênio GSD , o sistema é dano no nível molecular . Não porque se come muito açúcar, não porque o estilo de vida — mas porque uma enzima ou proteína transportadora específica não existe ou funciona pela metade. Como resultado: glicogênio acumula-se excessivamente no fígado/músculos como 'lixo químico' — ou, por outro lado, não pode ser construído diretamente. Não é diabetes. Não é hipoglicemia comum. É uma falha de máquina bioquímica congênita — e há mais de 16 tipos de GSD que são cientificamente reconhecidos, cada um causado por uma mutação genética específica.
Por que bebês parecem saudáveis na primeira semana — e então colapsam após 4 horas de jejum?
A maioria dos GSD especialmente tipos I, III, VI, IX apresenta os primeiros sintomas entre os dias 3 e 2 meses de vida — quando a reserva de glicose pré-natal se esgota . Bebês recém-nascidos ainda podem se alimentar de glicose da placenta; mas assim que nascem, eles dependem de glicogênio do fígado. No GSD tipo I von Gierke , a enzima glukose-6-fosfatoase não existe. Portanto, mesmo que o fígado esteja cheio de glicogênio, não pode liberar glicose para o fluxo sanguíneo . Como resultado: hipoglicemia grave em 3-4 horas sem leite. Seus sintomas? Lassidão, fraqueza, azulão, convulsões — e se não for tratado imediatamente, coma metabólico em menos de 24 horas. Uma pesquisa no Hospital Kuala Lumpur 2022 registrou 7 casos de GSD-I não diagnosticados até os 18 meses — todos entraram no ICU por hipoglicemia sem causa conhecida.
Por que os testes de sangue comuns frequentemente falham em detectar GSD — e quais testes são reais que devem ser feitos?
Muitos médicos suspeitam de infecção ou epilepsia quando bebês têm convulsões sem febre. Os testes de sangue básicos glicose, eletrolitos, função do fígado mostram hipoglicemia + aumento de látex + triglicerídeos altos — mas isso também ocorre em sepsis ou falha hepática aguda. O sinal único do GSD? Hipoglicemia com látex alto E ASIDOSA METABÓLICA QUE NÃO MELHORA após a administração de glicose intravenosa . Se a glicose IV melhorar a hipoglicemia — é provável que não seja GSD. Se não, é um forte sinal de falha da via de gliconeogênese. O teste conclusivo: análise de enzimas em biópsia de fígado padrão ouro , ou sequenciamento do gen G6PC para GSD-Ia , AGL para GSD-III , ou PHKA2 para GSD-IX . Agora, o painel de genética do GSD pode ser realizado em 10 dias — sem biópsia — no Centro Genético Nacional UKM.
O GSD pode ser 'curado'? E por que a dieta rica em milho não é mito — mas protocolo diário obrigatório?
Nenhum tratamento genético ou remédio mágico — mas o GSD pode ser controlado completamente com gerenciamento nutricional preciso. Para o GSD-I, a estratégia principal é prevenir hipoglicemia mantendo a corrente de glicose contínua . Como? Comer a cada 2-3 horas durante o dia e à noite — usando amido cru de milho não cozido UCCS . Esse amido é digerido lentamente, fornecendo glicose estável por 6-8 horas. Uma pesquisa longitudinal de 20 anos na Singapura mostrou que 94% dos pacientes com GSD-I que seguiram UCCS desde bebês nunca tiveram coma metabólico — e alcançaram crescimento e desempenho acadêmico normais. Importante: o UCCS não é suplemento , mas medicamento. A dose deve ser calculada com base no peso e idade — e deve ser iniciada sob supervisão de um especialista em metabolismo , não por sugestão de uma tia no WhatsApp.
Por que o GSD não é apenas 'doença de criança' — e quais riscos de longo prazo são frequentemente ignorados?
Muitos pensam que o GSD 'desaparece' quando o filho cresce. Erro grave. Sem gerenciamento a vida inteira, complicações surgem: fígado gorduroso progressivo → sirose → câncer de fígado risco 10x maior aos 30 anos ; aumento de ácido úrico → gota & cálculos renais; hiperlipidemia → aterosclerose precoce. No GSD-II doença de Pompe , a fraqueza muscular progressiva pode levar ao insuficiência respiratória — e agora há uma enzima substituta alglucosidase alfa que pode prolongar a vida em 10-15 anos se for administrada antes dos sintomas clínicos. O mais crítico: a triagem neonatal para GSD ainda não foi incluída no Programa de Triagem Neonatal Nacional da Malásia — embora possa ser detectada por espectrometria de massa tandem no terceiro dia de sangue de corda. Na Alemanha e no Japão, essa triagem reduziu a mortalidade de GSD-I em 78% nas últimas décadas.
Como as famílias podem agir — e por que 'esperar por sintomas claros' é a decisão mais arriscada?
Se há história familiar de GSD, ou o bebê apresenta: 1 hepatomegalia sem causa clara, 2 hipoglicemia recorrente antes dos 6 meses, 3 atraso do desenvolvimento motor associado à fraqueza muscular — não espere . Consulte imediatamente um Genetista ou um Metabolista de Crianças. Na Malásia, o Hospital Universitário de Malaca PPUM , o Hospital Sultanah Bahiyah Alor Setar, e o Instituto de Medicina Genética da USM oferecem consultas e testes genéticos licenciados. Há também o Clube de Apoio ao GSD da Malásia — uma comunidade de pais que compartilha protocolos de alimentação, dicas de UCCS e apoio emocional 24 horas. Lembre-se: o GSD não é uma punição. É um diagnóstico — e um diagnóstico preciso, no momento certo, é o início de uma vida cheia, ativa e longa. Como disse um paciente de 32 anos com GSD-III de Johor: ‘Eu não vivo com GSD. Eu vivo mesmo com GSD — e eu já corri duas maratonas.’
---
Rreferência: Doença de armazenamento de glicogênio — Wikipédia https://en.wikipedia.org/wiki/Glycogen storage disease