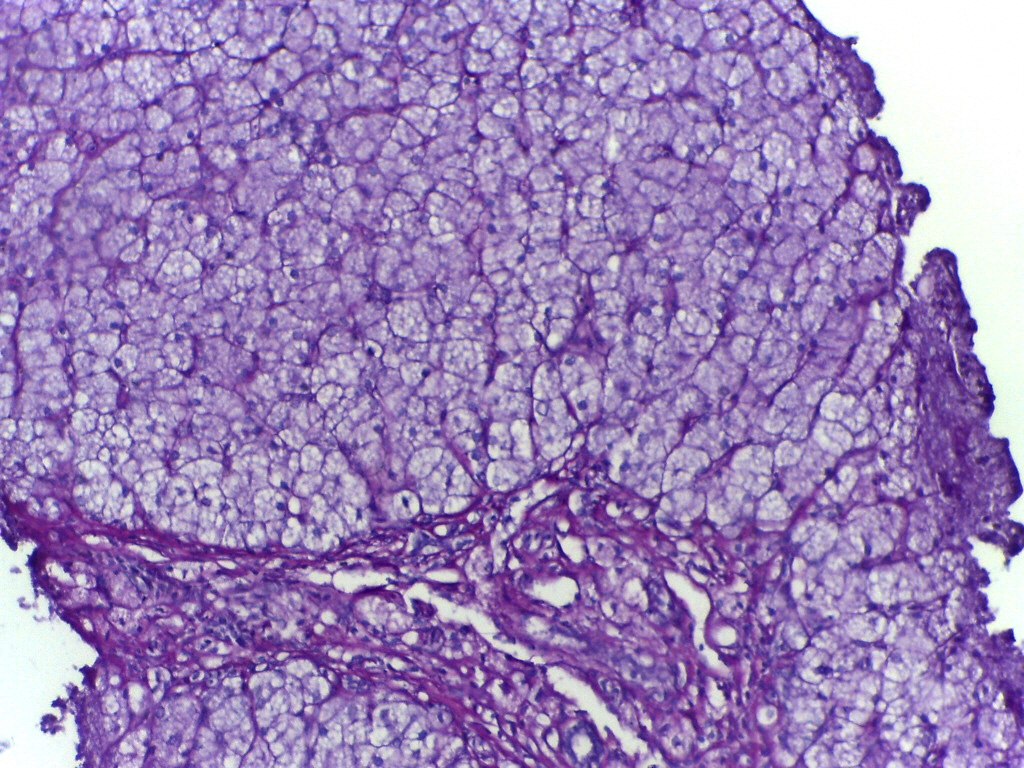
What is actually 'glycogen storage disease' — and why is it not just a 'sugar problem'?
Imagine glycogen like a backup battery in the body: stored mainly in the liver and muscles, then released as glucose when energy is needed — for example during overnight sleep, short fasts, or physical activity. In patients with Glycogen Storage Disease (GSD), this system
fails at the molecular level. Not because of eating too much sugar, not because of lifestyle — but because a specific enzyme or transporter protein
does not exist at all, or only functions partially. As a result: excess glycogen accumulates in the liver/muscles like 'chemical waste' — or alternatively, cannot be built at all. It is not diabetes. Not regular hypoglycemia. It is a failure of the biochemical machine — and there are more than 16 scientifically recognized types of GSD, each caused by a specific gene mutation.
Why do babies appear healthy in the first week — and then suddenly collapse after a 4-hour fast?
Most GSD (especially types I, III, VI, IX) show their first symptoms between day 3 to month 2 of life —
when prenatal glucose stores run out. Newborns can still rely on glucose from the placenta; but once born, they depend on liver glycogen. In type I GSD (von Gierke), the glucose-6-phosphatase enzyme is absent. Therefore, even though the liver is full of glycogen, it
cannot release glucose into the bloodstream. As a result: severe hypoglycemia within 3–4 hours without milk. Symptoms? Weakness, lethargy, cyanosis, seizures — and if not treated immediately, a metabolic coma within less than 24 hours. A study at Kuala Lumpur Hospital (2022) recorded 7 cases of GSD-I not diagnosed until 18 months — all had entered ICU due to undiagnosed hypoglycemia.
Why do routine blood tests often fail to detect GSD — and what tests should be done?
Many doctors suspect infection or epilepsy when a baby has a seizure without fever. Basic blood tests (glucose, electrolytes, liver function) do show hypoglycemia + increased lactate + high triglycerides — but these also occur in sepsis or acute liver failure. Unique indicators of GSD?
Hypoglycemia with high lactate AND metabolic acidosis THAT DOES NOT IMPROVE after intravenous glucose administration. If IV glucose quickly improves hypoglycemia — it is likely not GSD. If not, it is a strong indicator of failure in the gluconeogenesis pathway. Confirmatory test: enzyme analysis in a liver biopsy (gold standard), or gene sequencing of
G6PC (for GSD-Ia),
AGL (for GSD-III), or
PHKA2 (for GSD-IX). Now, GSD genetic panel tests can be conducted in 10 days — without a biopsy — at the National Genetic Center, UKM.
Can GSD be 'cured'? And why is a high corn diet not a myth — but a daily protocol must be followed?
There is no genetic treatment or magic drug — but GSD
can be fully controlled with precise nutritional management. For GSD-I, the main strategy is
preventing hypoglycemia by maintaining a continuous glucose supply. The way to do this: eat every 2–3 hours during the day and night — using
raw uncooked cornstarch (UCCS). This starch is slowly digested, providing stable glucose for 6–8 hours. A 20-year longitudinal study in Singapore showed that 94% of GSD-I patients who followed UCCS since infancy never experienced a metabolic coma — and achieved normal growth and academic performance. Important: UCCS
is not a supplement, but a medicine. Dosage must be calculated based on weight and age — and
must be started under the supervision of a metabolism specialist, not based on advice from a WhatsApp auntie.
Why is GSD not just a 'childhood disease' — and what long-term risks are often ignored?
Many think GSD 'disappears' when the child grows up. That is a big mistake. Without lifelong management, complications arise: progressive fatty liver → cirrhosis → liver cancer (risk 10 times higher in the 30s); increased uric acid → gout and kidney stones; hyperlipidemia → early atherosclerosis. In GSD-II (Pompe disease), progressive muscle weakness can lead to respiratory failure — and now there is an enzyme replacement (alglucosidase alfa) that can extend life by 10–15 years if given before clinical symptoms. Most critical:
neonatal screening for GSD is not included in Malaysia's National Neonatal Screening Program — although it can be detected via tandem mass spectrometry on day 3 of heel prick blood. In Germany and Japan, this screening has reduced GSD-I mortality by 78% in the last decade.
How can families act — and why is 'waiting for clear symptoms' the most risky decision?
If there is a family history of GSD, or the baby shows: (1) hepatomegaly without a clear cause, (2) recurrent hypoglycemia before 6 months of age, (3) motor development delay accompanied by muscle weakness —
do not wait. Immediately refer to a Geneticist or Pediatric Metabolism Specialist. In Malaysia, the University of Malaya Medical Centre (PPUM), Sultanah Bahiyah Alor Setar Hospital, and USM Institute of Genetic Medicine provide licensed consultations and genetic testing. There is also the Malaysian GSD Support Group — a parent community sharing food protocols, UCCS tips, and 24-hour emotional support. Remember: GSD is not a punishment. It is a diagnosis — and an accurate diagnosis at the right time is the beginning of a full, active, and long life. As one 32-year-old GSD-III patient from Johor said: *‘I am not living with GSD. I am living
despite GSD — and I have already run two marathons.’*
---
Reference: Glycogen storage disease — Wikipedia
Why This Child Can't Fast for More Than 4 Hours — Even Though He's Only 2 Years Old?. Behind the cute and calm face, his body holds a 'time bomb' of carbohydrates: glycogen that cannot be broken down. No obvious symptoms at birth — but hunger for 4 hours can trigger seizures, coma, or sudden death. This is not a myth. This is a rare disease affecting 1 in 20,000 babies — and 9 out of 10 cases are never diagnosed correctly before the age of 5.. What is actually 'glycogen storage disease' — and why is it not just a 'sugar problem'?
Imagine glycogen like a backup battery in the body: stored mainly in the liver and muscles, then released as glucose when energy is needed — for example during overnight sleep, short fasts, or physical activity. In patients with Glycogen Storage Disease GSD , this system fails at the molecular level . Not because of eating too much sugar, not because of lifestyle — but because a specific enzyme or transporter protein does not exist at all , or only functions partially. As a result: excess glycogen accumulates in the liver/muscles like 'chemical waste' — or alternatively, cannot be built at all. It is not diabetes. Not regular hypoglycemia. It is a failure of the biochemical machine — and there are more than 16 scientifically recognized types of GSD, each caused by a specific gene mutation.
Why do babies appear healthy in the first week — and then suddenly collapse after a 4-hour fast?
Most GSD especially types I, III, VI, IX show their first symptoms between day 3 to month 2 of life — when prenatal glucose stores run out . Newborns can still rely on glucose from the placenta; but once born, they depend on liver glycogen. In type I GSD von Gierke , the glucose-6-phosphatase enzyme is absent. Therefore, even though the liver is full of glycogen, it cannot release glucose into the bloodstream . As a result: severe hypoglycemia within 3–4 hours without milk. Symptoms? Weakness, lethargy, cyanosis, seizures — and if not treated immediately, a metabolic coma within less than 24 hours. A study at Kuala Lumpur Hospital 2022 recorded 7 cases of GSD-I not diagnosed until 18 months — all had entered ICU due to undiagnosed hypoglycemia.
Why do routine blood tests often fail to detect GSD — and what tests should be done?
Many doctors suspect infection or epilepsy when a baby has a seizure without fever. Basic blood tests glucose, electrolytes, liver function do show hypoglycemia + increased lactate + high triglycerides — but these also occur in sepsis or acute liver failure. Unique indicators of GSD? Hypoglycemia with high lactate AND metabolic acidosis THAT DOES NOT IMPROVE after intravenous glucose administration . If IV glucose quickly improves hypoglycemia — it is likely not GSD. If not, it is a strong indicator of failure in the gluconeogenesis pathway. Confirmatory test: enzyme analysis in a liver biopsy gold standard , or gene sequencing of G6PC for GSD-Ia , AGL for GSD-III , or PHKA2 for GSD-IX . Now, GSD genetic panel tests can be conducted in 10 days — without a biopsy — at the National Genetic Center, UKM.
Can GSD be 'cured'? And why is a high corn diet not a myth — but a daily protocol must be followed?
There is no genetic treatment or magic drug — but GSD can be fully controlled with precise nutritional management. For GSD-I, the main strategy is preventing hypoglycemia by maintaining a continuous glucose supply . The way to do this: eat every 2–3 hours during the day and night — using raw uncooked cornstarch UCCS . This starch is slowly digested, providing stable glucose for 6–8 hours. A 20-year longitudinal study in Singapore showed that 94% of GSD-I patients who followed UCCS since infancy never experienced a metabolic coma — and achieved normal growth and academic performance. Important: UCCS is not a supplement , but a medicine. Dosage must be calculated based on weight and age — and must be started under the supervision of a metabolism specialist , not based on advice from a WhatsApp auntie.
Why is GSD not just a 'childhood disease' — and what long-term risks are often ignored?
Many think GSD 'disappears' when the child grows up. That is a big mistake. Without lifelong management, complications arise: progressive fatty liver → cirrhosis → liver cancer risk 10 times higher in the 30s ; increased uric acid → gout and kidney stones; hyperlipidemia → early atherosclerosis. In GSD-II Pompe disease , progressive muscle weakness can lead to respiratory failure — and now there is an enzyme replacement alglucosidase alfa that can extend life by 10–15 years if given before clinical symptoms. Most critical: neonatal screening for GSD is not included in Malaysia's National Neonatal Screening Program — although it can be detected via tandem mass spectrometry on day 3 of heel prick blood. In Germany and Japan, this screening has reduced GSD-I mortality by 78% in the last decade.
How can families act — and why is 'waiting for clear symptoms' the most risky decision?
If there is a family history of GSD, or the baby shows: 1 hepatomegaly without a clear cause, 2 recurrent hypoglycemia before 6 months of age, 3 motor development delay accompanied by muscle weakness — do not wait . Immediately refer to a Geneticist or Pediatric Metabolism Specialist. In Malaysia, the University of Malaya Medical Centre PPUM , Sultanah Bahiyah Alor Setar Hospital, and USM Institute of Genetic Medicine provide licensed consultations and genetic testing. There is also the Malaysian GSD Support Group — a parent community sharing food protocols, UCCS tips, and 24-hour emotional support. Remember: GSD is not a punishment. It is a diagnosis — and an accurate diagnosis at the right time is the beginning of a full, active, and long life. As one 32-year-old GSD-III patient from Johor said: ‘I am not living with GSD. I am living despite GSD — and I have already run two marathons.’
---
Reference: Glycogen storage disease — Wikipedia https://en.wikipedia.org/wiki/Glycogen storage disease